
What to Know About Sickle Cell Disease
An expert explains what sickle cell disease is, how it impacts the body, and how the disease can be treated.
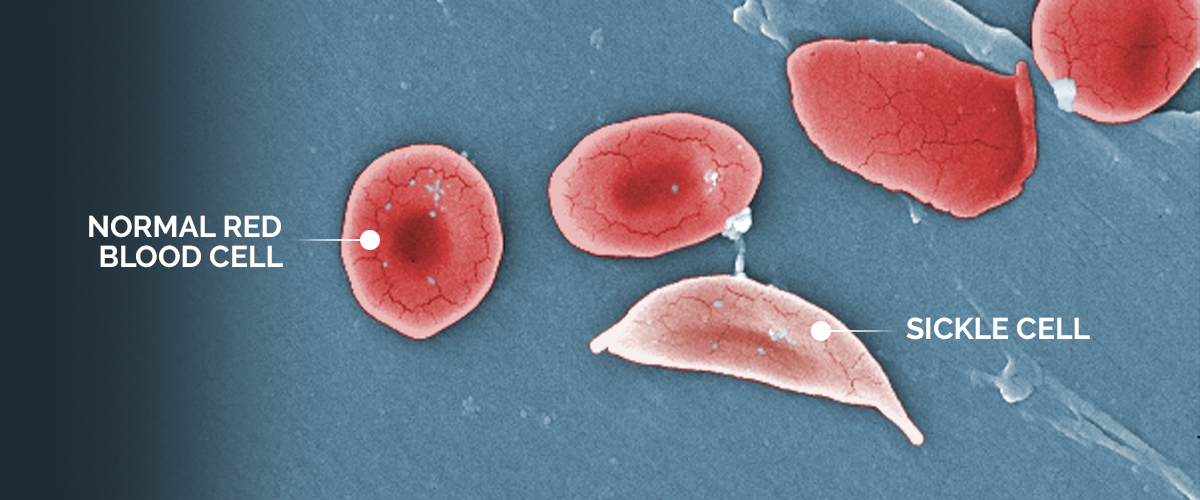
An image shows the structural difference between normal red blood cells and a sickle cell red blood cell.
Sickle cell disease is a group of genetic blood disorders that affects approximately 20 million people worldwide and an estimated 100,000 in the United States, making it the most common form of an inherited blood disorder in the country, according to the National Heart, Lung, and Blood Institute.
“Sickle cell disease is a result of abnormal production of red blood cells,” says Dr. Monica Bhatia, director of the Pediatric Stem Cell Transplant Program at NewYork-Presbyterian Morgan Stanley Children’s Hospital. “Red blood cells are usually circular and move easily throughout the blood vessels. But in patients with the disease, they are crescent, or sickle-shaped.”
Dr. Monica Bhatia
Although anyone can inherit sickle cell disease, it disproportionately impacts communities of color. According to the Centers for Disease Control and Prevention, sickle cell disease occurs in one out of every 365 Black or African American births and one out of every 16,300 Hispanic American births. Plus, about one in 13 Black or African American people are born with the sickle cell trait.
We asked Dr. Bhatia, who is also an associate professor of pediatrics at the Columbia University Vagelos College of Physicians and Surgeons, to share additional insights about sickle cell disease, including how it impacts the body and recent advancements that make it easier to treat the disease.
How does the sickle shape of the cells affect people with sickle cell disease?
Dr. Bhatia: The sickle-shaped red blood cells affect hemoglobin, which is the protein that carries oxygen throughout the body. Due to their irregular shape, the cells become clogged in the vessels. And when the red blood cells get stuck in vessels, they decrease the oxygen to organs.
This causes pain throughout the body that can take days to resolve. Some patients can manage the episodes at home with pain medicines, while others need to be in the hospital; these hospitalizations can be lengthy, up to two weeks in some cases. Among other complications, people with the condition can experience not only pain crises, but anemia, strokes, and organ damage.
How is the disease diagnosed?
There are prenatal and newborn screening options available. Newborns can be diagnosed with the disease by about two weeks of age. Symptoms begin to develop anywhere after four to six months. One of the early symptoms in newborns is dactylitis, which is swelling of the hands and feet. This is very painful and sometimes requires admission to the hospital for pain control.
How do the health challenges associated with sickle cell disease impact patients’ lives?
As children get older, they can have pain crisis episodes not only in their hands and feet, but in their back, shoulders, and chest. Some children are unfortunately at risk for strokes and can have significant cognitive decline, as well as physical dysfunctions such as vision loss and liver and kidney problems, to name a few.
Often in pediatric care settings, these patients miss school because of their pain crises. I have had children report to me that they are missing one to two weeks a month. This then has a downstream effect in terms of what and how much they can achieve. Many children have had to repeat a grade.
Patients with sickle cell disease also have shortened lifespans. Most patients with the most severe form of the disease have median life expectancies of about 50 years of age, which is 20 to 30 years less than those who do not have sickle cell disease. Studies have shown that up to 50% of people with sickle cell disease are unable to maintain consistent jobs due to the sequelae of the disease and have to go on disability.
There are multiple clinical trials and studies right now that are looking at modifying the genes of a patient with sickle cell disease. The nice thing about gene editing therapies is that you are using your own cells, so the chance of rejection is minimal.
Dr. Monica Bhatia
What treatments exist for sickle cell disease?
Treatments can include medicines and blood transfusions. Medicines are given to patients to prevent the sickling of red blood cells, reduce and treat pain crises, reduce the risk of infection, and prevent other complications.
Most hematologists recommend that pediatric patients go on a medicine called hydroxyurea at about nine months of age. The purpose of hydroxyurea is to increase the amount of fetal hemoglobin, which is produced in utero. Fetal hemoglobin has a higher oxygen affinity, so it is able to effectively carry oxygen to vital organs. Some children do very well with hydroxyurea, and if started early, it may even be able to eliminate pain crises.
Patients are also put on penicillin prophylaxis, which started in the 1970s and has decreased the risk of infections. By using penicillin prophylaxis, we have essentially eliminated the risk of people dying from the disease very early in life, but still have patients dying in their 40s and 50s.
There are other supportive treatments such as chronic transfusions, which help with diluting the sickle cells but are usually used to prevent further strokes in patients who have had strokes previously.
Is there a cure?
Patients can receive bone marrow or stem cell transplants and be cured, but this approach is risky and used in severe cases. First, a patient would need a donor who is a full match, typically a sibling, but only 15% of patients with sickle cell disease have a sibling who is a match and also doesn’t have the disease themselves.
If the patient does have a match, cure rates are about 90% to 95%. But for people who do not have a sibling who is a full match, gene therapy is ideal, especially in patients who have multiple crises.
There are multiple clinical trials and studies right now that are looking at modifying the genes of a patient with sickle cell disease. The nice thing about gene editing therapies is that you are using your own cells, so the chance of rejection is minimal. It helps reduce the chances of a complication known as graft versus host disease, which is an immunological process in which a donor’s cells attack the organs of the recipient and, in severe cases, can cause damage to the organ.
What Is Gene Therapy?
Gene therapy involves replacing or adding normal genes for missing or defective ones to potentially provide a cure for people without a well-matched donor. Additionally, new strategies called gene-editing can be used to modify the function of genes.
Dr. Markus Mapara, director of the Adult Bone Marrow Transplantation and Cell Therapy Program, is leading several of the groundbreaking trials at NewYork-Presbyterian/Columbia University Irving Medical Center that offer a potential new treatment for sickle cell disease using these different gene therapy approaches.
One treatment involves using a patient’s own stem cells to introduce new, genetically engineered hemoglobin. They then produce red blood cells that are not as likely to sickle. “You cannot overstate the potential impact of this new therapy,” says Dr. Mapara. “People with sickle cell disease live in constant fear of the next pain crisis. This treatment could give people with this disease their life back.”
Other adult and pediatric trials, which both Dr. Mapara and Dr. Bhatia are involved in, use different gene-editing approaches to reprogram a patient’s stem cells to produce fetal hemoglobin, which helps improve oxygen flow throughout the bloodstream.
Learn more about clinical trials at NewYork-Presbyterian.
Monica Bhatia, M.D., is the director of the Pediatric Stem Cell Transplant Program at NewYork Presbyterian Morgan Stanley Children’s Hospital and associate professor of pediatrics at the Columbia University Vagelos College of Physicians and Surgeons. Dr. Bhatia specializes in the care of children with noncancerous blood disorders who may benefit from bone marrow transplantation, especially those with sickle cell anemia. She and her colleagues are working to make bone marrow transplants more available to patients and to reduce the side effects and complications of the treatment without compromising its effectiveness.