
A Teen Finds Hope in a Breakthrough Sickle Cell Therapy
Jonathan Lubin’s young life was spent in and out of hospitals because of his sickle cell disease. But a new gene-editing therapy offered him the possibility of a life without pain crises.

It’s a crisp fall Saturday, and 16-year-old Jonathan Lubin dribbles around a court in a game of pickup basketball with friends. He evades a steal, goes for a hook shot, and makes the basket. He high-fives a teammate as his mom, Fabienne Desir, and dad, JR Lubin, cheer him on from the sidelines.
What seems like a typical day in the life of a teenager is a quiet triumph for Jonathan. Most of his young life has been spent in and out of hospitals because of sickle cell disease, a genetic blood disorder that causes the production of red blood cells that can become sickle-shaped instead of circular. Because of their unusual shape, these cells can get stuck and cause blockage in the blood vessels, known as vaso-occlusion. This prevents the flow of oxygen to the body, leading to pain, organ damage, anemia, or stroke.
For Jonathan, living with the disease meant suffering from chronic, sudden bouts of pain that required care in a medical facility. These pain crises, or vaso-occlusive crises (VOCs), often required hospitalization. With few treatment options suitable for him, in the fall of 2020, Jonathan, then 12, became one of the youngest patients to enroll in a clinical trial of exagamglogene autotemcel, or what is now known as Casgevy, a gene-editing therapy that has since been approved by the FDA for the treatment of sickle cell disease in patients 12 years and older who have recurrent VOCs. Jonathan’s own stem cells were used to help him treat the disease.
It was a long and arduous process, but it helped bring Jonathan the hope of a life without pain crises.
A Young Life Lived in Pain
Jonathan was diagnosed with sickle cell disease at 2 weeks old after getting tested, as both parents were carriers of the trait. Although symptoms don’t typically start until 4 to 6 months old, his started at 2 months, when his fingers and toes became so swollen that he cried at the slightest touch. At 9 months old, he was rushed to the hospital after refusing to eat and crying for hours; tests revealed he had experienced his first pain crisis. “It was so hard to watch because I didn’t know how to calm him down,” Fabienne says. “As a mother, to not be able to do anything was unbearable.”
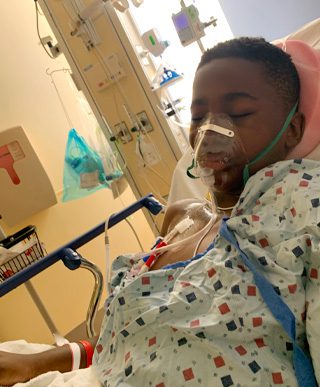
Jonathan’s pain crises required frequent hospitalizations.
As Jonathan grew older, his symptoms became more severe; anything from playing outside to jumping in a pool could trigger a pain crisis. He seldom went more than two or three months without a trip to the emergency room. “We always had a ‘go bag’ ready because you never knew when something would happen,” JR says. “We would take him to daycare or a birthday party, and then get a call that he was having a pain crisis. We’d grab the bag and get him to the hospital.” There were too many missed days of school, birthday parties, and pool parties to count.
There were also constant fevers, infections, skin ulcers, and organ damage. Jonathan had to have his spleen and gall bladder removed because of complications from his sickle cell disease. And when he was 10, just a few weeks before he planned to try out for the basketball team, he woke up with excruciating pain in his legs. He had developed avascular necrosis in his left hip, a condition in which bone tissue starts to decay because of a lack of blood flow. He could barely walk, let alone run or jump. “If somebody touched any part of my leg, it would hurt really bad,” Jonathan says. “Not being able to walk felt like the worst thing in the world because it’s something you do every day, it’s a normal thing. But I was literally bedridden.”
One potential treatment for some people with sickle cell disease is a bone marrow transplant from a healthy, matched related donor, often a sibling. A half-matched donor or a matched unrelated donor might also be possible. However, there is the risk of developing graft-versus-host disease, which is when donor immune cells recognize a host’s tissue as foreign and start to attack it.
With Jonathan being an only child, and with no good matches for him in the donor registry, his options were limited. So when the family heard that a clinical trial for a new sickle cell disease therapy was happening at NewYork-Presbyterian Morgan Stanley Children’s Hospital, where he was getting his treatment, they saw it as a ray of hope. “Over the course of Jonathan’s life, we realized how much of a roller coaster he’d been on health-wise,” JR says. “We knew we had to see what else was out there.”
Learning About a Different Approach
In November 2020, Jonathan enrolled in the clinical trial of Casgevy, a gene-editing therapy that uses CRISPR-Cas9 technology to edit a patient’s own stem cells before reintroducing them back to the body to help treat disease. For sickle cell patients, it is used to help them produce fetal hemoglobin, a type of hemoglobin that is produced during development and early life. In the first few months after birth, it is mostly replaced by adult hemoglobin — or, for those with sickle cell disease, by sickle hemoglobin. Hemoglobin is a protein in red blood cells that helps carry oxygen to the body.
Under the CRISPR-Cas9 approach, blood stem cells that have been harvested from a patient are sent to a manufacturing facility to edit a part of the BCL11A gene, which serves as an “off switch” to the production of fetal hemoglobin in red blood cells. The edited blood stem cells are given back to the same patient as a blood stem cell transplantation so that they can repopulate the bone marrow and enter the bloodstream. As a result of the gene editing, the transplanted blood stem cells help turn the production of fetal hemoglobin back on, thereby preventing the red blood cells from sickling.
Dr. Monica Bhatia
“The beauty of a gene therapy approach is that we basically take the donor out of the equation. There’s no risk of graft-versus-host disease or the potential rejection of the graft,” says Dr. Markus Mapara, director of the Bone Marrow Transplantation and Cell Therapy Program at NewYork-Presbyterian/Columbia University Irving Medical Center, who has led multiple clinical gene-therapy trials on adult patients. “In my eyes, this has the potential to have a major impact on patients with sickle cell disease. Results have been very encouraging, but it will be necessary to closely monitor the patients to ensure that the treatment success is durable, and to rule out that the genetic modification may lead to unintended damage,” says Dr. Mapara.
Undergoing the treatment can also be a long and arduous process, so Jonathan’s care team had to prepare the family. “The process is quite lengthy and is a big commitment on the family’s part,” says Dr. Monica Bhatia, director of the Pediatric Stem Cell Transplant Program at NewYork-Presbyterian Morgan Stanley Children’s Hospital, who was an investigator on the trial Jonathan was involved in and oversaw his care. “But as long as patients continue to produce high amounts of fetal hemoglobin, then there is a lot of promise for this new treatment.”
Jonathan’s Treatment Journey
From start to finish, it took about a year for Jonathan to complete his treatment, and it wasn’t without its trying moments.
The preparation included a bevy of tests and blood transfusions to ensure that he would be healthy enough for the collection and reception of his blood stem cells. After his stem cells were collected, it took several weeks for them to be edited and tested. When it was nearing time to receive his cells, he was admitted to the hospital to undergo four days of high-dose chemotherapy, which was necessary to clear cells from the bone marrow so that the gene-edited stem cells could engraft properly. But the chemotherapy led to a severe case of mucositis, an inflammation of the mouth and gastrointestinal tract. He developed painful mouth sores and couldn’t eat or talk.
“They told us what to expect, but it felt very different when you were living it,” JR says. “He was missing out on school, activities, day-to-day life, he had a tube coming out of his neck — it was difficult to see him going through all that.”

Jonathan, center, with his mom, Fabienne, and dad, JR. After completing gene-editing therapy in 2021, Jonathan can worry less about pain crises.
In October 2021, two weeks after receiving the infusion of his modified stem cells, things started to turn around. Jonathan recovered from the mucositis, and his blood cell counts improved. His appetite started to return, as did his energy level. “Jonathan really tried to engage in his care. I encouraged him to establish as much of a routine as possible, to get up every day, take his meds, do physical therapy, do homework,” Dr. Bhatia says. “People who tend to stay on a schedule do better, and Jonathan really tried to do that.”
In a little less than six weeks, he was cleared to leave the hospital. Weekly checkups turned into checkups once every three months. Now, Jonathan is down to once every six months, and he hasn’t had a pain crisis in over two years.
His care team will follow his progress for many years to come, but for now, Jonathan is simply enjoying living life without having to worry about pain crises. He jumped into a pool this summer. He plays drums for his church band. And for his 15th birthday, he and JR took a trip to Paris.
For Fabienne and JR, not trying to predict when the next hospitalization will be is still an unusual feeling. “It was hard at first, because for 13 years I was always waiting for the other shoe to drop,” Fabienne says. “It’s good to see him enjoying his life without me being worried.”
“We’re in a position where we can take a step back and say, ‘You know what? He’s got it,’” adds JR. “He’s been doing all the things he wants, and he’s been doing them fearlessly.”